Related Subjects:
|Iron deficiency Anaemia
|Haemolytic anaemia
|Macrocytic anaemia
|Megaloblastic anaemia
|Microcytic anaemia
|Myelodysplasia
|Myelofibrosis
The most important first step in the management of Essential Thrombocythaemia (ET) 🩸 is to confirm the diagnosis and exclude other myeloid neoplasms which may mimic ET (e.g. prefibrotic primary myelofibrosis, masked polycythaemia vera, chronic myeloid leukaemia, refractory anaemia with ring sideroblasts and thrombocytosis). Always involve haematology early.
📖 About
- 📈 Uncontrolled increase in platelet production from clonal haematopoiesis.
- 🧬 Classified as a myeloproliferative neoplasm (MPN).
- 🤝 Shares features with polycythaemia vera (PV) and primary myelofibrosis (PMF).
- ⚖️ Indolent course with median survival >20 years; risk of thrombosis, bleeding, and transformation increases with age and JAK2 mutation.
🧬 Aetiology
- Driver mutations: JAK2 V617F (~55–65%), CALR (~20–25%), MPL (~5%).
- Triple-negative ET (~10–15%) is rarer, often requires bone marrow to exclude prefibrotic PMF; generally lower thrombosis risk but may have inferior outcomes if misclassified.
📏 Definition / Diagnostic Criteria
- Sustained platelet count ≥450 × 10⁹/L (persistent ≥3 months recommended in some cases).
- Presence of JAK2, CALR, or MPL mutation (peripheral blood PCR preferred).
- If no mutation → exclude reactive thrombocytosis and other MPNs (bone marrow essential in higher-risk or unexplained cases).
- Bone marrow: proliferation mainly of megakaryocytic lineage with enlarged, mature megakaryocytes (hyperlobulated nuclei), loose clusters; no/minimal reticulin fibrosis (grade 0–1); exclude other myeloid neoplasms.
🩺 Clinical Features
- 🤕 Microvascular symptoms: headaches, erythromelalgia (burning pain/redness in extremities), visual disturbances, dizziness.
- ❤️ Constitutional symptoms, palpitations.
- 🩸 Thrombosis: arterial (stroke, MI), venous (DVT/PE, splanchnic vein thrombosis e.g. Budd–Chiari).
- ⚠️ Paradoxical bleeding: especially with extreme thrombocytosis (>1000–1500 × 10⁹/L) due to acquired von Willebrand factor (vWF) deficiency.
⚠️ Complications
- Progression to post-ET myelofibrosis (~10–20% lifetime risk).
- Transformation to acute myeloid leukaemia (~1–5%, higher with hydroxyurea long-term or prior thrombosis).
- Haemorrhage (acquired vWD), major thrombosis (main cause of morbidity).
🧪 Investigations
- Normal CRP, ferritin, iron studies to exclude reactive causes.
- Platelets often >600–1000 × 10⁹/L; blood film shows giant/abnormal platelets.
- Haematinics may be low if chronic bleeding.
- Driver mutation testing (JAK2/CALR/MPL); BCR-ABL1 to exclude CML.
- Bone marrow biopsy: key to confirm ET vs prefibrotic PMF (recommended in most cases per modern practice).
🔍 Differential of High Platelets
- Reactive: infection/inflammation, iron deficiency, malignancy, post-splenectomy, trauma/surgery.
- Other MPNs: PV (masked), prefibrotic PMF, CML.
- Rare: MDS with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T).
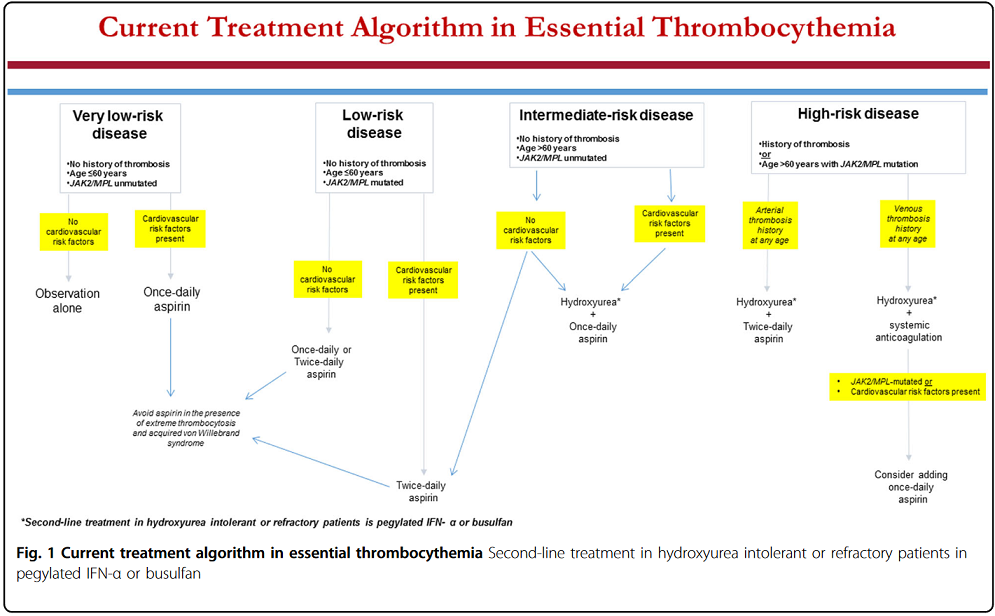
💊 Management (risk-stratified; per BSH consensus & 2024 international updates)
- 🧓 Risk stratification: Very low (≤60y, no thrombosis, JAK2-neg); Low (≤60y, no thrombosis, JAK2-pos); Intermediate (>60y, no thrombosis, JAK2-neg); High (thrombosis history or >60y + JAK2-pos). CV risk factors and CALR/MPL status influence.
- 🩺 Low/very low risk → low-dose aspirin (75–100 mg daily; twice-daily in low-risk per some experts). Avoid in extreme thrombocytosis with acquired vWD (test vWF activity/multimer pattern first).
- 💉 Cytoreduction: Hydroxyurea first-line in UK for high-risk (or intermediate if symptomatic); pegylated interferon-α (e.g. ropeginterferon) preferred in young/pregnant or as alternative (disease-modifying potential); anagrelide second-line; busulfan rarely for elderly.
- 📊 Target: Platelets <400 × 10⁹/L in high-risk; symptom control in lower-risk. Monitor for progression.
- Special: Pregnancy → interferon preferred; splanchnic thrombosis → anticoagulation + cytoreduction.
Key References: BSH 2025 guideline on thrombocytosis without JAK2/CALR/MPL (link); Tefferi 2024 update on ET diagnosis/risk/management (Am J Hematol); BSH 2014 diagnostic criteria modification. No specific NICE TA; refer to CKS platelets-abnormal counts.
Cases - Essential Thrombocythaemia (ET)
- Case 1 - Incidental Thrombocytosis:
A 62-year-old woman has a routine FBC for hypertension follow-up. Platelets are 720 ×10⁹/L (persistently elevated), Hb and WCC are normal. She is asymptomatic, no splenomegaly. JAK2 mutation positive. Diagnosis: Essential Thrombocythaemia (low-risk, asymptomatic).
- Case 2 - Thrombotic Complication:
A 55-year-old man presents with sudden onset left-sided weakness. CT confirms an ischaemic stroke. Bloods show platelets 980 ×10⁹/L, Hb 13 g/dL, WCC 9 ×10⁹/L. He has a history of erythromelalgia. JAK2 V617F positive. Diagnosis: ET complicated by arterial thrombosis (high-risk).
- Case 3 - Bleeding in Extreme Thrombocytosis:
A 70-year-old woman presents with recurrent epistaxis and easy bruising. Platelets are 1,600 ×10⁹/L, Hb 12.5 g/dL, WCC 11 ×10⁹/L. Acquired von Willebrand factor deficiency confirmed. Diagnosis: ET with bleeding complication from acquired vWF deficiency.
Teaching Commentary 🧪
Essential Thrombocythaemia is a myeloproliferative neoplasm characterised by sustained thrombocytosis (>450 ×10⁹/L) due to clonal proliferation driven by JAK2, CALR, or MPL mutations. Patients may be asymptomatic or present with microvascular symptoms (erythromelalgia, headaches), thrombosis (arterial/venous), or bleeding at extreme counts (acquired vWD). Diagnosis requires mutation testing ± bone marrow to exclude mimics like prefibrotic PMF. Management is risk-stratified: aspirin for most (to prevent thrombosis); cytoreduction (hydroxyurea first-line in UK high-risk) to reduce events. Indolent course with excellent survival, but monitor for transformation. (BSH 2025 & Tefferi 2024)
