Related Subjects:
|Sickle Cell Disease
|Acute Chest Syndrome (Sickle Cell)
|Exchange Transfusion
🩸 Sickle Cell Disease (SCD) → Inform haematology immediately if a known HbSS/SC patient is admitted.
🔄 In SCD, red cells become “sickle-shaped” under hypoxic stress → vaso-occlusion, haemolysis, and multiorgan complications.
📖 About
- 🌍 Chronic haemolytic anaemia, mainly affecting people of Afro-Caribbean origin.
- ⚡ Most severe: HbSS (≈70% UK cases).
- 🟡 HbSC: milder but still crisis-prone.
- 🧬 HbAS (trait): usually asymptomatic, but gene carrier & protective against malaria.
- 🆘 Patients often carry a haemoglobinopathy alert card.
🧬 Aetiology
- 🧪 Point mutation: Valine replaces glutamic acid at position 6 of β-globin.
- 🔗 HbS polymerises at ↓ O₂ → rigid, sickled cells → vaso-occlusion + haemolysis.
- 🔍 Diagnosis: Hb electrophoresis or HPLC.
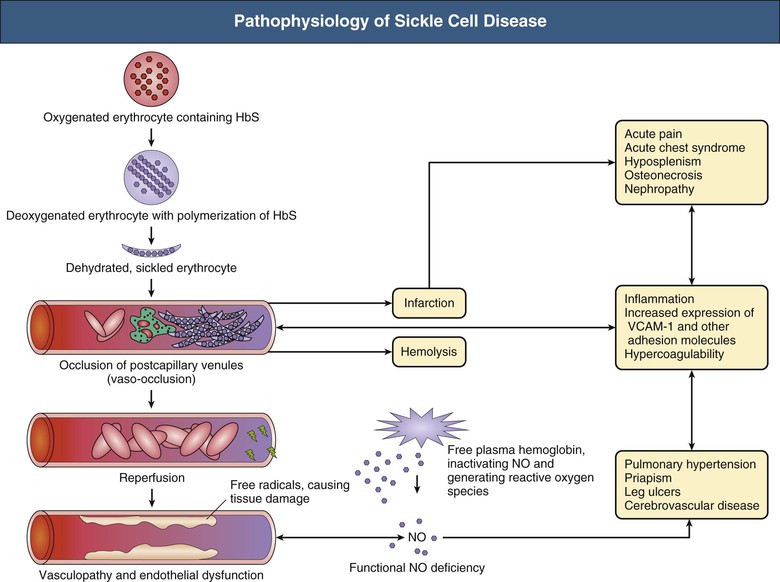
⚡ Precipitants of Sickling
- ⛰️ Deoxygenation (high altitude, hypoxia).
- 💧 Dehydration.
- 🤒 Infection, fever.
- 🤰 Pregnancy, 😰 stress, 🧪 acidosis.
🩺 Types of Haemoglobinopathy
- 🔴 HbSS: severe, anaemic, recurrent crises.
- 🟡 HbSC: milder, vaso-occlusion possible.
- ⚫ HbS/β-thalassaemia: resembles HbSS.
- 🟢 Trait (HbAS): asymptomatic carrier.
👩⚕️ Clinical Presentation
- 🌬️ Acute chest syndrome: fever, dyspnoea, chest pain, hypoxia, falling Hb → ICU risk.
- 🦴 Painful vaso-occlusive crisis: bone pain (esp. long bones, spine, dactylitis in children).
- 🪫 Splenic sequestration: sudden splenomegaly + hypovolaemia (esp. children).
- 🧠 Neurological: stroke from cerebral sickling.
- 🍆 Priapism: prolonged painful erection → urgent input.
- 🦠 Aplastic crisis: parvovirus B19 → profound anaemia.
- 💛 Cholecystitis: gallstones from chronic haemolysis → jaundice + RUQ pain.
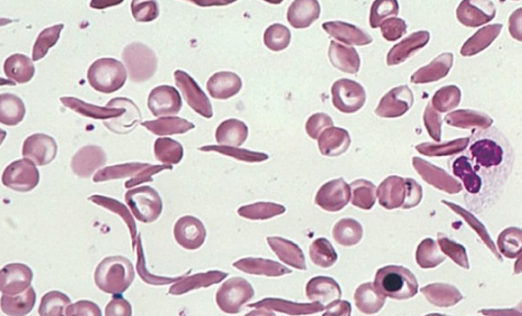
🚑 Complications
- 🌬️ Acute chest syndrome.
- 🛡️ Recurrent infections (autosplenectomy → pneumococcus, Salmonella osteomyelitis).
- 🦵 Avascular necrosis (femoral head common).
- 🩸 Renal papillary necrosis, haematuria.
- 🍆 Priapism → risk of erectile dysfunction.
- 🩹 Leg ulcers, hepatosplenomegaly, pulmonary fibrosis.
🔎 Investigations
- 🩸 FBC → anaemia, ↑ reticulocytes.
- 🔬 Blood film → sickled cells, target cells.
- 🦠 Sepsis screen if febrile.
- 🌬️ ABG & sats → hypoxia (chest crisis vs PE).
- 🖼️ CXR → consolidation/effusion.
- 🧠 CT/MRI brain → if neurological deficit/stroke.
- 🧪 U&E, LFTs, LDH, bilirubin, haptoglobin → haemolysis & organ function.
💊 Management of Acute Crisis
- 🛑 ABCDE + senior review early.
- 💧 Hydration: IV saline (avoid overload).
- 💊 Pain relief: Morphine within 30 mins, titrate + NSAID + paracetamol. Add laxatives.
- 🌬️ O₂ if sats <95%.
- 🦠 Antibiotics if fever/sepsis (cover pneumococcus/Salmonella).
- 💉 Consider exchange transfusion for: acute chest, stroke, priapism, multi-organ failure (target HbS <30%).
📅 Long-Term Management
- 💊 Hydroxyurea → ↑ HbF, ↓ crises.
- 💉 Vaccinations: pneumococcus, Hib, meningococcus.
- 💊 Prophylactic penicillin in children.
- 🧬 Bone marrow transplantation → curative in selected patients.
📚 Exam / OSCE Pearls
– 🚨 Sickle crises are medical emergencies → treat pain + fluids promptly.
– 🤒 Always rule out infection.
– 🌬️ Chest crisis: O₂ + antibiotics + consider exchange transfusion.
– 🍆 Ask about priapism, 🧠 neuro symptoms, 🪫 splenic sequestration in any acute admission.
📖 References
Cases - Sickle Cell Disease
- Case 1 - Vaso-occlusive crisis 🔥: A 19-year-old man with known HbSS genotype presents with sudden severe generalised bone pain, especially in his long bones and back. He is febrile (38.1°C) and tachycardic. FBC: Hb 7.8 g/dL, WCC 15.0 ×10⁹/L. Diagnosis: acute painful crisis in sickle cell disease. Managed with opioid analgesia, IV fluids, oxygen, and infection screen with empiric antibiotics.
- Case 2 - Acute chest syndrome 🫁: A 24-year-old woman with sickle cell disease presents with pleuritic chest pain, fever, cough, and increasing shortness of breath. CXR: new bilateral infiltrates. ABG: hypoxia. Diagnosis: acute chest syndrome (vaso-occlusion in pulmonary vasculature ± infection). Managed with oxygen, antibiotics, analgesia, and exchange transfusion if hypoxia worsens.
- Case 3 - Stroke in childhood 🧠: A 10-year-old boy with sickle cell disease is brought with sudden right-sided weakness and slurred speech. CT head: ischaemic stroke. Bloods: Hb 6.9 g/dL. Diagnosis: ischaemic stroke due to sickle cell vaso-occlusion. Managed acutely with exchange transfusion and long-term with chronic transfusion programme and hydroxycarbamide.
Teaching Point 🩺: Sickle cell disease is an inherited haemoglobinopathy (HbSS) leading to haemolysis and vaso-occlusion. Key complications include: painful crises, acute chest syndrome, stroke, splenic sequestration, infections, chronic organ damage. Hydroxycarbamide, vaccination, prophylactic antibiotics, and transfusion programmes improve survival.
