
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Systemic Amyloid in Pathology
Amyloid refers to a diverse group of protein aggregates that are pathologically deposited in tissues and organs. These aggregates have a characteristic beta-sheet structure that leads to their insolubility and resistance to proteolysis. Amyloid deposits are associated with various diseases, collectively known as amyloidosis.
Characteristics of Amyloid
- Beta-sheet Structure :
- Amyloid fibrils are composed of proteins that misfold into beta-sheet-rich structures.
- This structure is responsible for the fibrils' stability and resistance to degradation.
- Congo Red Staining :
- Amyloid deposits exhibit apple-green birefringence under polarized light when stained with Congo red dye.
- Electron Microscopy :
- Amyloid fibrils appear as non-branching, linear structures with a diameter of 7-10 nm under electron microscopy.
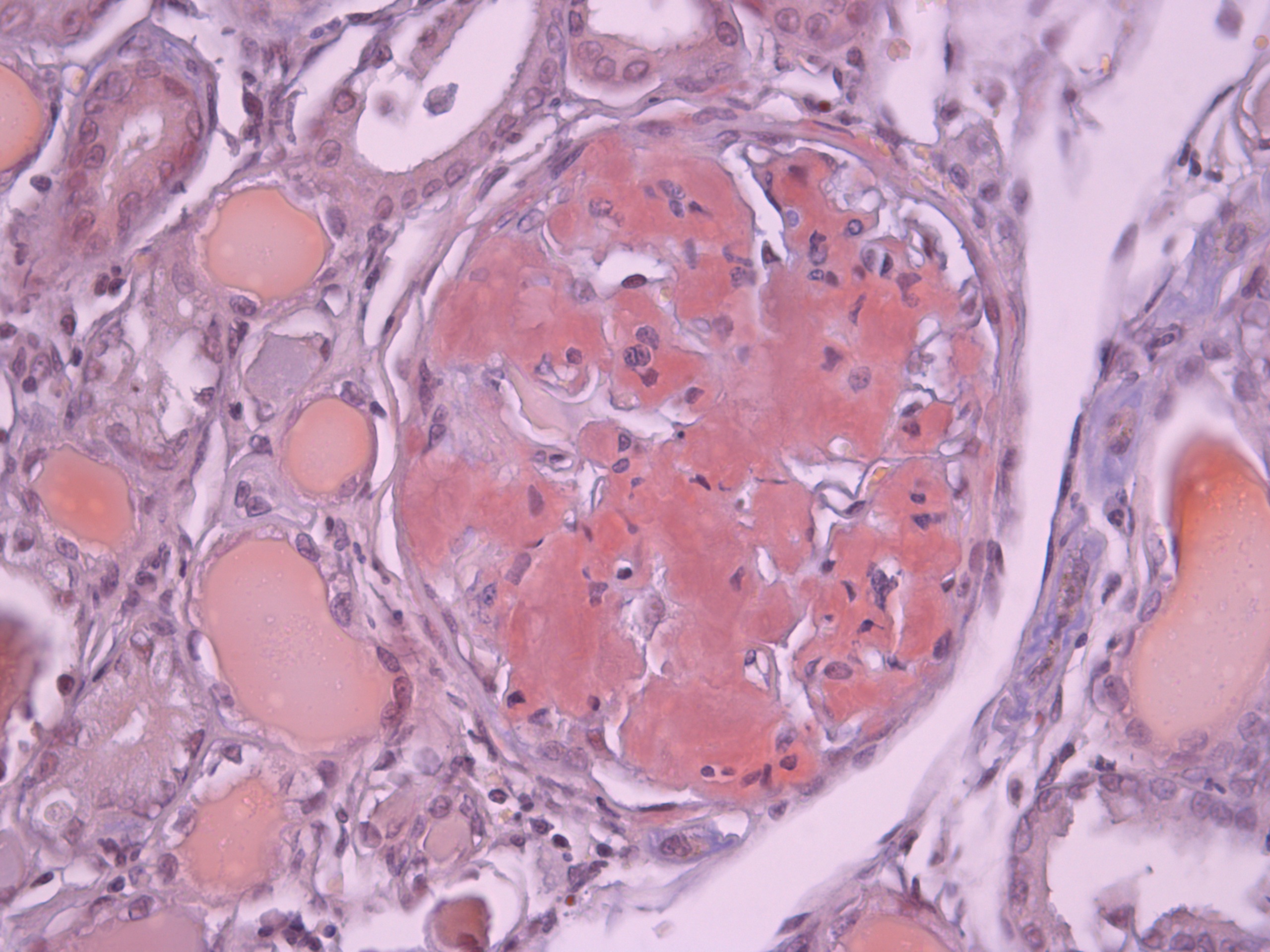
Types of Amyloid Proteins
- AL Amyloid (Amyloid Light Chain) :
- Derived from immunoglobulin light chains, often associated with plasma cell dyscrasias like multiple myeloma.
- AA Amyloid (Amyloid A) :
- Derived from serum amyloid A protein, associated with chronic inflammatory conditions like rheumatoid arthritis.
- ATTR Amyloid (Transthyretin) :
- Derived from the transthyretin protein, associated with familial amyloid polyneuropathy and senile systemic amyloidosis.
- ABeta Amyloid :
- Derived from amyloid precursor protein (APP), associated with Alzheimer's disease.
- Abeta2M Amyloid (Beta-2 Microglobulin) :
- Associated with long-term haemodialysis.
Pathogenesis of Amyloidosis
- Protein Misfolding :
- Genetic mutations or environmental factors can cause proteins to misfold into beta-sheet structures.
- Misfolded proteins aggregate to form amyloid fibrils.
- Deposition :
- Amyloid fibrils are deposited extracellularly in tissues and organs.
- Deposits disrupt normal tissue structure and function.
- Organ Dysfunction :
- Accumulation of amyloid deposits leads to progressive organ dysfunction and failure.
Clinical Manifestations
- Renal Involvement :
- Proteinuria and nephrotic syndrome.
- Progressive renal failure.
- Cardiac Involvement :
- Restrictive cardiomyopathy.
- Arrhythmias and heart failure.
- Neurological Involvement :
- Peripheral neuropathy.
- Autonomic dysfunction.
- Gastrointestinal Involvement :
- Malabsorption and weight loss.
- Hepatomegaly and liver dysfunction.
- Musculoskeletal Involvement :
- Carpal tunnel syndrome.
- Muscle weakness and wasting.
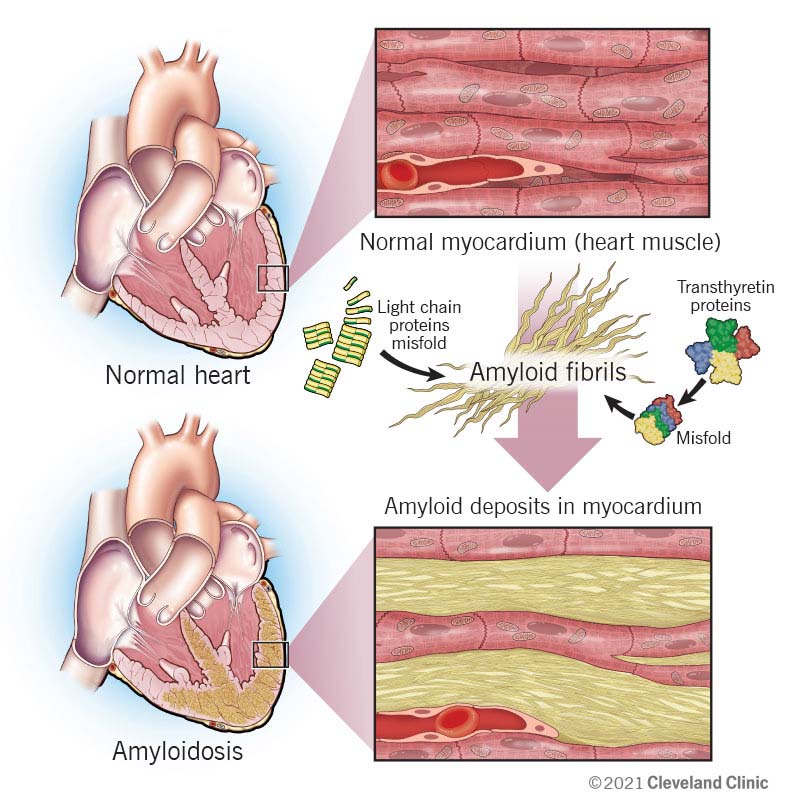
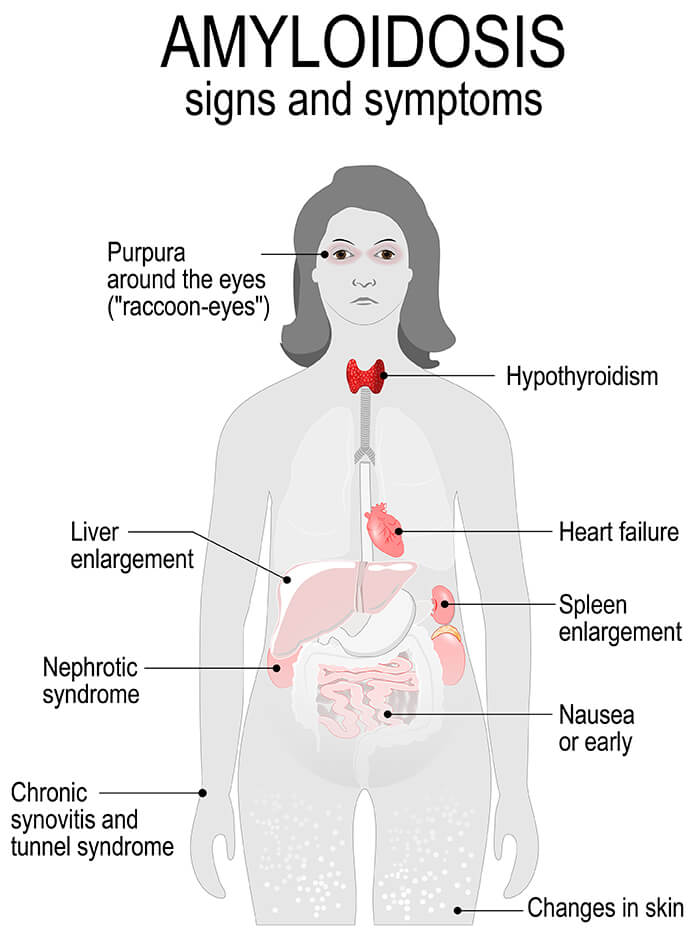
Diagnosis of Amyloidosis
- Biopsy :
- Tissue biopsy (e.g., abdominal fat pad, rectal, or organ biopsy) with Congo red staining.
- Immunohistochemistry :
- Identifies the specific type of amyloid protein.
- Serum and Urine Protein Electrophoresis :
- Detects monoclonal light chains in AL amyloidosis.
- Genetic Testing :
- Identifies mutations in familial amyloidosis.
- Imaging :
- Cardiac MRI and echocardiography for cardiac involvement.
Treatment of Amyloidosis
- Targeting Underlying Disease :
- Treating the underlying condition (e.g., multiple myeloma, chronic inflammation).
- Chemotherapy :
- Used for AL amyloidosis to reduce the production of amyloid light chains.
- Organ Transplantation :
- Kidney, liver, or heart transplantation in cases of severe organ involvement.
- Supportive Care :
- Management of symptoms and complications (e.g., diuretics for heart failure, pain management for neuropathy).
Summary
Amyloid is a pathological protein aggregate associated with a variety of diseases, known collectively as amyloidosis. These aggregates have a characteristic beta-sheet structure, and their deposition in tissues leads to organ dysfunction. Amyloidosis can involve different types of amyloid proteins, including AL, AA, ATTR, ABeta, and Abeta2M. Diagnosis involves tissue biopsy, immunohistochemistry, and imaging studies. Treatment focuses on addressing the underlying condition, reducing amyloid production, and managing symptoms and organ dysfunction.