
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Wilson disease
Related Subjects: |Chronic liver disease |Cirrhosis |Alkaline phosphatase (ALP) |Liver Function Tests |Ascites Assessment and Management |Budd-Chiari syndrome |Autoimmune Hepatitis |Primary Biliary Cirrhosis |Primary Sclerosing Cholangitis |Wilson disease |Hereditary Haemochromatosis |Alpha-1 Antitrypsin (AAT) deficiency |Non alcoholic steatohepatitis (NASH) |Spontaneous Bacterial Peritonitis |Alcoholism and Alcoholic Liver Disease
Wilson's disease accounts for 6–12% of all acute liver failure cases referred for emergency liver transplantation. It should be suspected in any patient with unexplained liver disease or movement disorders of uncertain cause. Age alone must not be used to exclude the diagnosis.
📖 About
- Wilson’s disease is an autosomal recessive disorder of copper metabolism.
- Prevalence ~1 in 30,000 live births; carrier frequency ~1 in 90.
- Untreated → progressive copper accumulation in the liver, brain, cornea, and kidneys, leading to cirrhosis, neuropsychiatric decline, and early death.
- Early recognition and treatment are life-saving.
🧬 Aetiology
- Mutations in the ATP7B gene on chromosome 13 → defective copper-transporting ATPase.
- Defect prevents copper excretion into bile and incorporation into caeruloplasmin.
- Over 200 ATP7B mutations have been identified worldwide.
Kayser–Fleischer rings (copper in Descemet’s membrane of the cornea) are a hallmark feature. Best seen on slit-lamp exam, but not entirely specific.
🩺 Clinical Features
- Onset typically between ages 5–45.
- Hepatic: hepatitis, cirrhosis, hepatosplenomegaly, jaundice, acute liver failure.
- Haematological: Coombs-negative haemolytic anaemia (free copper damages RBC membranes).
- Neurological: dysarthria, dystonia, rigidity, tremor, choreoathetosis (due to basal ganglia copper).
- Ophthalmic: Kayser–Fleischer rings, sunflower cataracts (posterior lens surface).
- Psychiatric: irritability, poor impulse control, depression, psychosis.
- Musculoskeletal: osteoporosis, rickets from chronic disease.
🔍 Investigations
- Liver function: ↑ ALT/AST, prolonged PT in advanced disease.
- Haematology: Coombs-negative haemolysis.
- Biochemical markers:
- ↓ Serum caeruloplasmin (<0.2 g/L; <0.1 g/L highly specific).
- ↑ Free serum copper (>1.6 µmol/L).
- ↑ 24-hr urinary copper (>1.6 µmol/24h).
- ↑ Hepatic copper content (>250 µg/g dry weight).
- Ophthalmology: Slit lamp for Kayser–Fleischer rings.
- Renal: Tubular dysfunction → proteinuria, glycosuria.
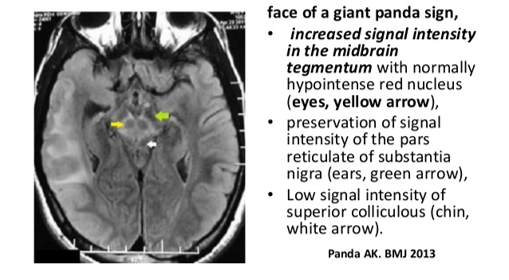
- MRI brain: basal ganglia/cerebellar changes, “giant panda sign”.
- Genetics: ATP7B mutation testing confirms but complicated by heterogeneity.
The combination of Kayser–Fleischer rings + low caeruloplasmin (<0.1 g/L) is diagnostic.
💊 Management
- Untreated Wilson’s disease is universally fatal.
- Copper chelation:
- D-penicillamine = first-line (but can cause lupus-like syndrome, nephrosis).
- Always give Pyridoxine (B6) with penicillamine.
- Trientine (triethylene tetramine) as alternative chelator.
- Zinc acetate: blocks intestinal copper absorption; safe for maintenance therapy.
- Dietary copper restriction (avoid shellfish, nuts, chocolate, mushrooms, liver).
- Liver transplantation: curative in fulminant hepatic failure or advanced cirrhosis.
📚 Reference
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery