
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Motor Neuron Disease (MND-ALS)
Related Subjects: |Neurological History taking |Motor Neuron Disease (MND-ALS) |Miller-Fisher syndrome |Guillain Barre Syndrome |Multifocal Motor Neuropathy with Conduction block |Multiple Sclerosis (MS) Demyelination |Inclusion Body Myositis |Cervical spondylosis |Anterior Spinal Cord syndrome |Central Spinal Cord syndrome |Brown-Sequard Spinal Cord syndrome |Spinal Cord Compression |Spinal Cord Haematoma |Spinal Cord Infarction
🧠 Motor Neurone Disease (MND) presents with generalized and bulbar weakness, muscle wasting, brisk reflexes, and no sensory loss. Eye and bladder function are typically spared. ⚡ There is no definitive diagnostic test - diagnosis relies on clinical signs involving both the brain and spinal cord.
ℹ️ About
- Also known as Amyotrophic Lateral Sclerosis (ALS), Charcot's disease, and Lou Gehrig's disease.
- ⏳ Diagnosis is often delayed, sometimes taking >16 months from symptom onset.
- Initial symptoms may be vague, e.g., fatigue or weakness.
How Common Is It? 📊
- Incidence: 1.8–2.2 per 100,000 population.
- Prevalence: 4.0–4.7 per 100,000 population in the UK.
- At any given time, ~2,000 individuals in England and Wales are affected.
Pathology 🔬
- Degeneration of motor giant pyramidal Betz cells (layer V, primary motor cortex).
- Loss of anterior horn cells in the spinal cord.
- Degeneration of cranial motor nuclei in the brainstem.
Aetiology / Genetics 🧬
- 🌍 Guam variant: linked with dementia & parkinsonism in the Chamorro people.
- SOD1 mutations implicated in familial ALS.
- Other mutations: RNA processing, axonal transport, cytoskeletal proteins.
- Familial ALS = ~10% of cases, usually autosomal dominant.
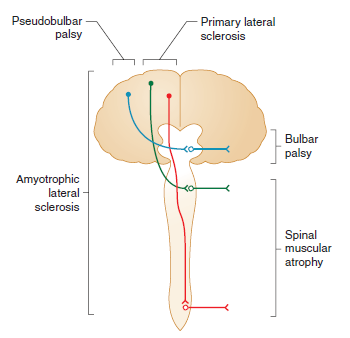
Forms of MND 🧾
- Progressive Muscular Atrophy: LMN predominant. ⏳ Survival 5–10 yrs.
- Progressive Bulbar Palsy: Tongue wasting, fasciculations, spastic palate. ⏳ Survival 2–3 yrs.
- Primary Lateral Sclerosis: UMN predominant, symmetrical. 👍 Good prognosis.
- ALS: UMN + LMN features. ⏳ Survival 3–4 yrs.
- MND-Dementia: Associated with FTD (frontotemporal dementia).
Clinical Features ⚠️
- 💪 Subtle weakness → progresses to wasting & fasciculations (esp. hands).
- 🌙 Painful nocturnal cramps (thighs common).
- Mixed UMN + LMN signs: weakness + brisk reflexes + extensor plantar.
- 🙅♂️ No significant sensory loss; eye movements & sphincters spared.
- 👅 Bulbar involvement: wasted fibrillating tongue, dysarthria, dysphagia.
- ⚖️ Weight loss due to dysphagia & wasting.
- “Dropped head” & wasted hands appearance.
El Escorial Criteria 📋
- Clinically Definite ALS: UMN + LMN signs in ≥3 regions (bulbar, cervical, thoracic, lumbar).
- Clinically Probable ALS: UMN + LMN in 2 regions, UMN above LMN.
- Clinically Possible ALS: UMN + LMN in 1 region only.
Differential Diagnosis 🔍
- MMN with conduction block (check GM-1 antibodies).
- Cervical spondylotic myelopathy, foramen magnum lesions (MRI).
- CIDP, Inclusion Body Myositis.
- Myasthenia Gravis (Tensilon, antibodies, EMG).
- Kennedy’s syndrome (X-linked).
- B12 deficiency, Lyme disease, malignancy.
Investigations 🧪
- FBC, U&E, TFT, CK, syphilis serology.
- CSF: may show mild ↑ protein.
- Autoantibody panel (exclude mimics).
- MRI brain/cervical spine → rule out mimics (syrinx, compression, vascular).
- EMG: fasciculations + fibrillation (LMN loss).
- Nerve conduction: often normal until late.
Management 🩺
- 👩⚕️ Multidisciplinary Support: Physio, OT, SALT, dietician, palliative input.
- 💊 Riluzole 50mg bd: Glutamate antagonist → extends survival by ~2–3 months.
- 💧 Anticholinergics: Glycopyrrolate, amitriptyline, or hyoscine for drooling.
- 🦵 Baclofen / Diazepam: Spasticity control.
- 🥤 Feeding support: NG, PEG, or RIG feeding when bulbar involvement advances.
- 😴 NIV (BiPAP): Improves sleep, reduces fatigue, ↑ survival by ~6 months.
- 🗣️ Speech synthesizers: Assist communication.
- 💊 Quinine, Carbamazepine, Gabapentin → muscle cramps.
- 🙂 SSRIs or mood stabilisers for depression & emotional lability.
Prognosis 📉
- Median survival: 2–3 yrs from diagnosis.
- ~25% survive ≥5 yrs.
References 📚
Cases - Motor Neurone Disease (MND)
- Case 1 - Amyotrophic Lateral Sclerosis (ALS, Mixed UMN & LMN) ⚡: A 58-year-old man presents with progressive weakness in his right hand, muscle fasciculations, and stiffness in his legs. Exam: wasting of hand intrinsic muscles, brisk reflexes, extensor plantar responses. No sensory loss. Diagnosis: ALS (most common form of MND). Management: Riluzole to modestly extend survival; physiotherapy; MDT input; NIV (non-invasive ventilation) for respiratory support.
- Case 2 - Progressive Bulbar Palsy 🗣️: A 62-year-old woman presents with slurred speech, difficulty swallowing, and choking on liquids. Exam: tongue wasting with fasciculations, brisk jaw jerk, nasal dysarthria. Diagnosis: Bulbar-onset MND (progressive bulbar palsy). Management: MDT care; speech and language therapy; gastrostomy for nutrition; NIV; symptomatic treatment of sialorrhoea.
- Case 3 - Primary Lateral Sclerosis (PLS, UMN-Predominant) 🧠: A 50-year-old man presents with progressive leg stiffness and spastic gait over 3 years. Exam: pyramidal weakness, brisk reflexes, extensor plantar responses, but no fasciculations or wasting. Sensation intact. Diagnosis: Primary lateral sclerosis (UMN-predominant MND variant). Management: Supportive - physiotherapy, spasticity control (baclofen, tizanidine), MDT monitoring.
Teaching Commentary 🧠
MND is a group of progressive neurodegenerative disorders of upper and lower motor neurones. - ALS: UMN + LMN signs, most common. - Bulbar Palsy: speech and swallowing first; poorer prognosis. - PLS: UMN-predominant; slower progression. - PMA (progressive muscular atrophy): LMN-predominant. Clues: weakness with fasciculations, mixed UMN/LMN signs, no sensory loss, progressive. Dx: clinical + EMG (denervation); MRI excludes mimics. Mx: Riluzole, multidisciplinary care, NIV, gastrostomy, palliative support. Prognosis: average survival 2–5 years (longer in PLS).
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery