Charcot Marie Tooth (CMT) disease
🧠 Age of onset is variable depending on subtype, penetrance, family history, and ascertainment bias. Most patients develop symptoms in childhood or early adulthood, but subtle features may go unnoticed until later in life. Always consider CMT in young patients with progressive distal weakness, foot deformities, and a positive family history.
ℹ️ About
- Charcot-Marie-Tooth (CMT) disease is the most common inherited peripheral neuropathy. It is a length-dependent, progressive motor and sensory neuropathy.
- Also known as Hereditary Sensorimotor Neuropathy (HSMN).
- Motor involvement (weakness, wasting, foot drop) tends to dominate, but sensory loss can be subtle or subclinical.
🌍 Epidemiology
- Prevalence: ~40 per 100,000 (≈ 1 in 2,500) → one of the most common inherited neurological disorders.
- Slightly more common in males 👨 than females 👩.
- Lower reported prevalence in African American populations.
🧬 Aetiology
- Most forms are autosomal dominant (AD), but autosomal recessive (AR) and X-linked (XLR) types also exist.
- Demyelinating forms (e.g. CMT1) are most common, but some are primarily axonal (CMT2).
- PMP22 gene duplication on chromosome 17 is the single most frequent mutation, producing abnormal myelin protein and slowed nerve conduction.
- In total, >50 genetic mutations identified (including GJB1, MFN2, MPZ, GDAP1).
🔗 Associations
- Ophthalmic: Retinitis pigmentosa (notably in CMT type 7).
- Musculoskeletal: Scoliosis, hip dysplasia.
🩺 Clinical Presentation
- Distal weakness & wasting: “glove-and-stocking” pattern with peroneal muscle wasting.
- Characteristic legs: Inverted “champagne bottle” appearance 🍾 due to calf wasting.
- Hand involvement: Claw hands ✋, impaired fine motor control.
- Foot deformities: Pes cavus (high arch) 🦶, hammer toes, or sometimes pes planus.
- Neurological findings: Foot drop, absent ankle jerks, reduced vibration and proprioception.
- Patients often recall difficulty running 🏃 in school sports.
- Thickened peripheral nerves (palpable ulnar or common peroneal) in some cases.
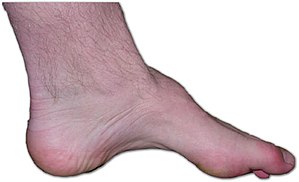
🖼️ Image
Example of CMT foot deformity: high arch, claw toes, and distal muscle wasting.
📑 Subtypes of CMT
| Type | Features |
|---|
| CMT Type I |
AD, demyelinating. Very slow nerve conduction (<38 m/s). Distal wasting, pes cavus, foot drop. Onset childhood/teens. |
| CMT Type II |
AD, axonal. Conduction velocity near-normal. Later onset (teens/adulthood), milder course. |
| CMT Type III (Dejerine-Sottas) |
AR, severe. Early childhood onset, profound weakness, sensory loss, enlarged nerves. CSF protein ↑ (>10 g/L). |
🧪 Investigations
- Genetic testing: Confirms subtype (PMP22 duplication commonest).
- Nerve conduction studies: Slowed in demyelinating types; relatively preserved in axonal forms.
- Electromyography (EMG): Myopathic vs neuropathic differentiation.
- CSF analysis: Protein ↑ in Dejerine-Sottas (CMT3).
- MRI spine/brain: Only if atypical features or to exclude other causes.
⚠️ Factors that Worsen Symptoms
- Emotional stress 😟
- Prolonged immobility 🛌
- Pregnancy 🤰 (progesterone-related worsening)
- Certain drugs 💊: vincristine, amiodarone, colchicine (iatrogenic neuropathy risk)
🛠️ Management
- Life expectancy: Normal in most cases, but progressive disability is common.
- Multidisciplinary care: Neurology, physiotherapy, orthotics, podiatry, and genetic counseling.
- Mobility: Ankle-foot orthoses for foot drop; wheelchair only in severe cases.
- Foot care: Daily inspection, avoid barefoot walking (risk of unnoticed injuries due to sensory loss).
- Genetic counseling: Important for family planning, given high penetrance of AD forms.
- Supportive devices: Hand splints, adaptive aids for fine motor skills.
- Respiratory support: Sleep apnoea screening and management if symptomatic.
🧾 Key Exam Pearls
✅ “Inverted champagne bottle legs” = classic CMT clue.
✅ Pes cavus + distal wasting + family history = consider CMT.
✅ Distinguish from diabetic neuropathy: CMT = early onset, family history, foot deformities.
✅ NCS: slowed velocity in demyelinating (CMT1), relatively normal in axonal (CMT2).
📚 References
